
Слєпов О.К.
Науковий керівник - Слєпов Олексій Костянтинович, доктор медичних наук, професор, Заслужений лікар України
телефон (044) 483-22-80
Завідувач клінічного відділення торакоабдомінальної хірургії вад розвитку у новонароджених і дітей різних вікових груп з ліжками урогінекології (45 ліжок) - Пономаренко Олексій Петрович, лікар вищої категорії, кандидат медичних наук
телефон (044) 483-62-28
Співробітники (науковий та лікарській склад відділення):
Сорока Василь Петрович - провідний науковий співробітник, кандидат медичних наук, Заслужений лікар України.
Джам Олег Петрович - дитячій хірург, вища категорія, кандидат медичних наук, науковий співробітник.
Мигур Михайло Юрійович - дитячий хірург, ІІ категорія, молодший науковий співробітник.
Пономаренко Олексій Петрович - завідувач клінічного відділення, лікар вищої категорії, кандидат медичних наук.
Палкіна Ірина Сергіївна - педіатр, лікар ендоскопіст, вища категорія, кандидат медичних наук.
Зайченко Оксана Миколаївна - лікар акушер-гінеколог, дитячий гінеколог, спеціаліст з ультразвукової діагностики, вища категорія.
Коцовський Володимир Васильович - дитячий хірург, ІІ категорія.
Пономаренко Максим Вікторович - дитячий уролог, вища категорія.
Скиба Олександр Степанович - дитячий хірург, дитячий ортопед-травматолог, дитячий уролог, І категорія.
Сердюк Андрій Сергійович - дитячий хірург.
У 1981 році було створене відділення торакоабдомінальної і судинної хірургії вад розвитку у дітей.
З 2006 року науковий підрозділ було перейменовано у відділення хірургічної корекції вад розвитку у дітей - очолює Заслужений лікар України, доктор медичних наук, професор Слєпов Олексій Костянтинович.
Клінічною базою наукового підрозділу є відділення торако-абдомінальної хірургії вад розвитку у новонароджених і дітей різних вікових груп з ліжками урогінекології. Відділення розраховане на 45 ліжок і розташоване на 5, 4 та 3 поверхах дитячого корпусу установи.
У відділенні функціонують:
операційний блок (дві операційні та супутні приміщення),
3 кабінети ендоскопічних досліджень, 2 перев’язочні кабінети,
2 маніпуляційних кабінети, 1 гінекологічна оглядова,
9 палат для хворих на 5 поверсі
та 6 палат на 4 поверсі,
2 їдальні та інше.
Відділення оснащено сучасним обладнанням: ендоксопами Olympus та Storz, лапароскопічною стійкою Olympus, діатермокоагуляторами (електроніж) з аргоновим ножем Martin, наркозними станціями Dragger, інструментарієм, сучасною апаратурою цілодобового моніторингу за станом життєвих показників пацієнтів, сучасним апаратом ультразвукової діагностики, мобільною рентген установкою.
Клініка дитячої хірургії у складі науково-клінічних підрозділів є унікальним, єдиним в Україні місцем де надається комплексна хірургічна допомога при вадах розвитку у дітей. Виключною особливістю ДУ «ІПАГ ім. акад. О.М. Лук’янової НАМН України» є те, що в стінах єдиного науково-лікувального закладу зібрані відділення планування сім’ї, медицини плода, акушерські клініки, відділення реанімації новонароджених та інтенсивної терапії і реанімації дитячих клінік, відділення хірургічної корекції вроджених вад розвитку у дітей, багатопрофільні соматичні відділення дитячих клінік. Це дає змогу рано виявляти (з 12 тижня вагітності), диспансерно спостерігати та планувати подальшу тактику ведення вагітності, пологів та спеціалізованого хірургічного лікування великої кількості вад розвитку у дітей. Налагоджено тісну співпрацю з усіма закладами системи охорони здоров’я України, а також з деякими клініками Бельгії, США, Польщі, Італії, Ізраїлю, особливо з дитячими обласними лікарнями м. Чернігова, м. Житомира, м. Вінниці та м. Миколаєва.
Основними напрямками науково-лікувальної роботи клініки дитячої хірургії у складі відділення хірургічної корекції природжених вад розвитку у дітей, та його клінічної бази - відділення торакоабдомінальної хірургії вад розвитку у новонароджених і дітей різних вікових груп з ліжками урогінекології, є надання хірургічної допомоги новонародженим і дітям різних вікових груп з вродженими вадами розвитку, а саме:
- Вроджені вади розвитку органів дихальної системи.
- Вроджені вади розвитку органів травної системи.
- Вроджені вади розвитку кістково-м’язової системи (діафрагмальна грижа, гастрошизис, омфалоцелє, тощо).
- Вроджені вади розвитку органів сечовидільної системи.
- Захворювання, що супроводжуються обструкцією дихальних шляхів, шлунково-кишкового тракту та сечовидільної системи.
- Пухлини та пухлиноподібні утворення грудної клітки, черевної порожнини, заочеревинного простору, м’яких тканин, тощо.
- Хірургічна патологія органів дихальної системи, органів травної системи, кістково-м’язової системи, сечовидільної системи, тощо.

Завідувач відділення, заслужений лікар України, д.м.н., проф., Слєпов О.К. із своїми співробітниками
Методи діагностики і лікування
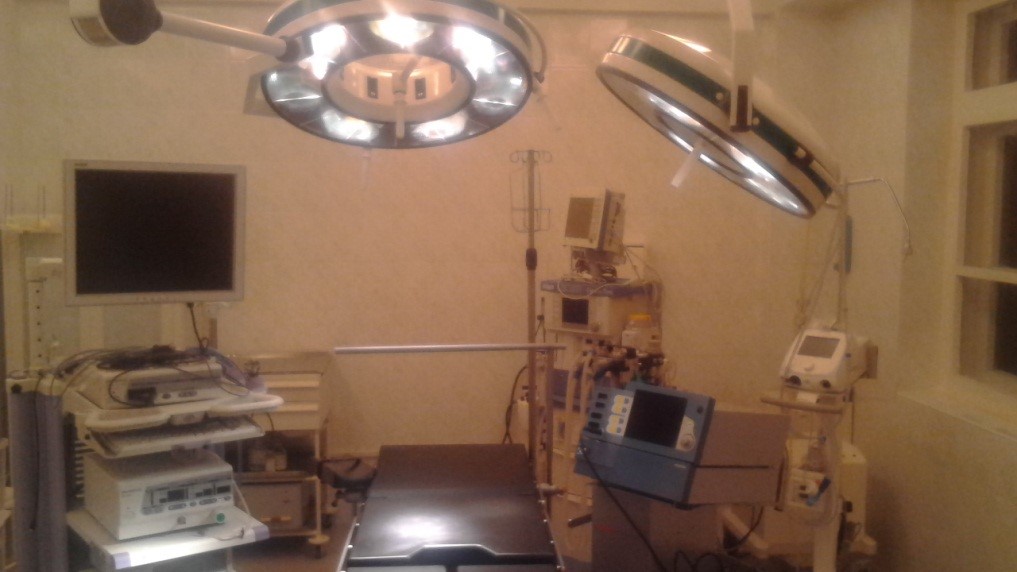 В клінічному відділенні застосовуються сучасні методики та підходи при лікуванні таких вад розвитку як атрезія стравоходу, діафрагмальна грижа, гастрошизис, омфалоцелє, об’ємні утворення грудної та черевної порожнин, межистіння та заочеревинного простору, а також м’яких тканин різних локалізацій, кишкова непрохідність (атрезія дванадцятипалої кишки, атрезія тонкої кишки, атрезія товстої кишки, хвороба Гіршпрунга, аноректальні атрезії, тощо).
В клінічному відділенні застосовуються сучасні методики та підходи при лікуванні таких вад розвитку як атрезія стравоходу, діафрагмальна грижа, гастрошизис, омфалоцелє, об’ємні утворення грудної та черевної порожнин, межистіння та заочеревинного простору, а також м’яких тканин різних локалізацій, кишкова непрохідність (атрезія дванадцятипалої кишки, атрезія тонкої кишки, атрезія товстої кишки, хвороба Гіршпрунга, аноректальні атрезії, тощо).
Розробляється та впроваджується складне, етапне хірургічне лікування вад розвитку грудної та черевної порожнин. При необхідності, діти отримують хірургічну допомогу з перших хвилин життя та спостерігаються до 18 років. Лікування новонароджених дітей з природженими вадами розвитку є об’єктом особливої уваги. Застосування сучасних методів діагностики та лікування виконується із застосуванням новітнього обладнання та згідно розроблених сучасних клінічних протоколів.
Для раннього виявлення патології плода застосовується скринінгова пренатальна діагностика. Після виявлення вади розвитку у плода консиліумом (генетик, дитячий хірург, анестезіолог, акушер-гінеколог) вирішується питання щодо подальшого ведення вагітності та диспансеризації вагітної і плода, визначаються терміни та спосіб родорозрішення. Пологи відбуваються в присутності дитячого хірурга й дитячого анестезіолога-реаніматолога, новонародженій дитині з перших секунд життя проводиться адекватна інтенсивна терапія, для подальшого лікування дитина переводиться у відділення дитячої реанімації, де продовжується передопераційна підготовка і паралельне дообстеження немовляти з метою виявлення супутніх вад розвитку або захворювань. Після чого проводиться хірургічна корекція вади розвитку (при гастрошизісі – в перші хвилини після народження, при інших захворюваннях – в ранньому неонатальному періоді).
Основні захворювання, на яких спеціалізується дитяча хірургічна клініка - атрезія стравоходу, діафрагмальна грижа, гастрошизис, омфалоцелє, кишкова непрохідність (Хв. Гіршпрунга, атрезія тонкої, атрезія товстої кишки), аноректальні вади розвитку, об'ємні утворення грудної порожнини та межистіння, черевної порожнини та заочеревинного простору, м'яких тканин, доліхоколон, урологічна патологія (гідронефроз, міхурово-сечовідний рефлюкс, гіпоспадія, фімоз, водянка яєчка, крипторхізм тощо), гінекологічна патологія (порушення циклу, кісти яєчників, маткові кровотечі, синехії, тощо), ахалазія кардії, грижа стравохідного отвору діафрагми, пілоростеноз, пупкові нориці, пупкові грижі, пахові грижі тощо. Також проводяться мініінвазивні хірургічні втручання і, в тому числі, лапароскопічні та ендоскопічні, пункційні біопсії печінки. Ці операції виконуються за допомогою сучасного обладнання, що дає можливість проводити як діагностичні так і лікувальні втручання у дітей різних вікових груп. При необхідності, також надається допомога і пацієнтам із сторонніми тілами шлунково-кишкового тракту та дихальних шляхів, а також з гострою хірургічною патологією.
Розробка та провадження медико-організаційних заходів, що включають профілактику, перинатальну діагностику, доопераційну стабілізацію, раннє хірургічне лікування дало можливість суттєво знизити післяопераційну летальність у новонароджених дітей з життєво-небезпечними вадами розвитку в ДУ «ІПАГ НАМН України».
Цей механізм забезпечується шляхом надання безперервної допомоги матері та дитині в одній лікувальній установі, починаючи з планування вагітності, та закінчуючи випискою додому здорової дитини та матері.
Вперше в світі було впроваджено тактику «Хірургії перших хвилин» при лікуванні гастрошизису у новонароджених дітей, що полягає в хірургічному втручанні через 10-20 хвилин після їх народження. При цьому вдалося підвищити виживання цих важких пацієнтів до 82,0 %.
При природженій діафрагмальній грижі, вперше в Україні, впроваджено нову тактику і стратегію діагностики та лікування цієї патології у плодів та новонароджених дітей. При цьому досягнуто збільшення виживання цих критичних хворих до 92 %, що є високим показником навіть для розвинених країн світу. Також значно зменшено післяопераційну летальність при атрезії стравоходу (до 7 %), при омфалоцелє – до 10 %, при природженій кишковій непрохідності – до 15 %.